妙啊!用擴(kuò)散模型生成蛋白質(zhì)結(jié)構(gòu),結(jié)果不輸天然蛋白質(zhì)|來自斯坦福&微軟
網(wǎng)友:未來5年用文本提示生成新抗體/新酶也不是夢吧
豐色 發(fā)自 凹非寺
量子位 | 公眾號(hào) QbitAI
沒想到,圖像生成領(lǐng)域的大明星——
擴(kuò)散模型,這么快就被用來做蛋白質(zhì)結(jié)構(gòu)生成了!
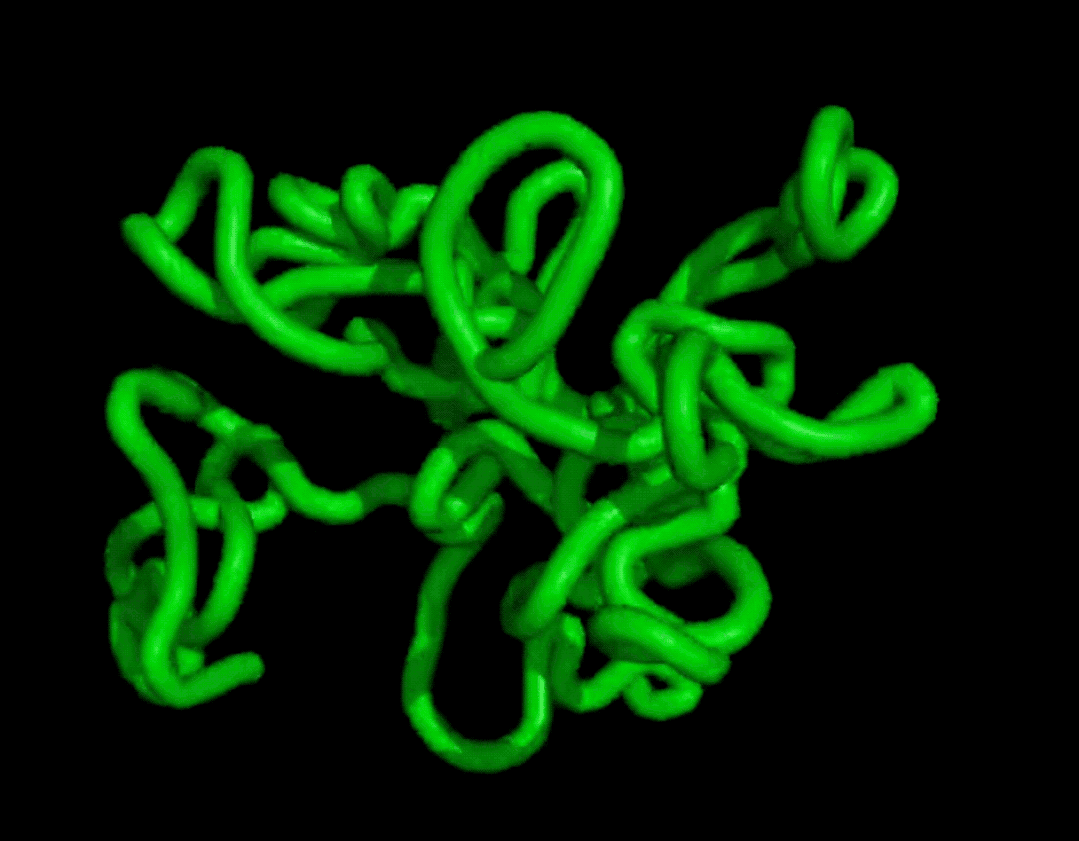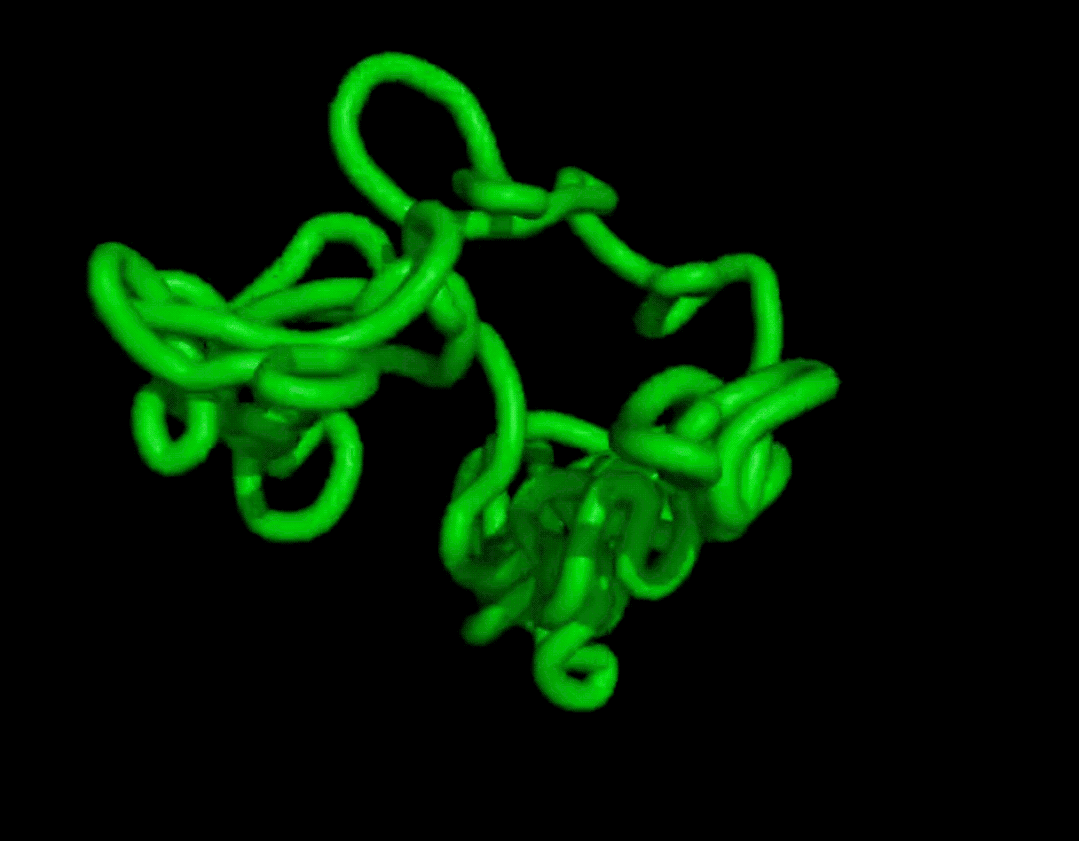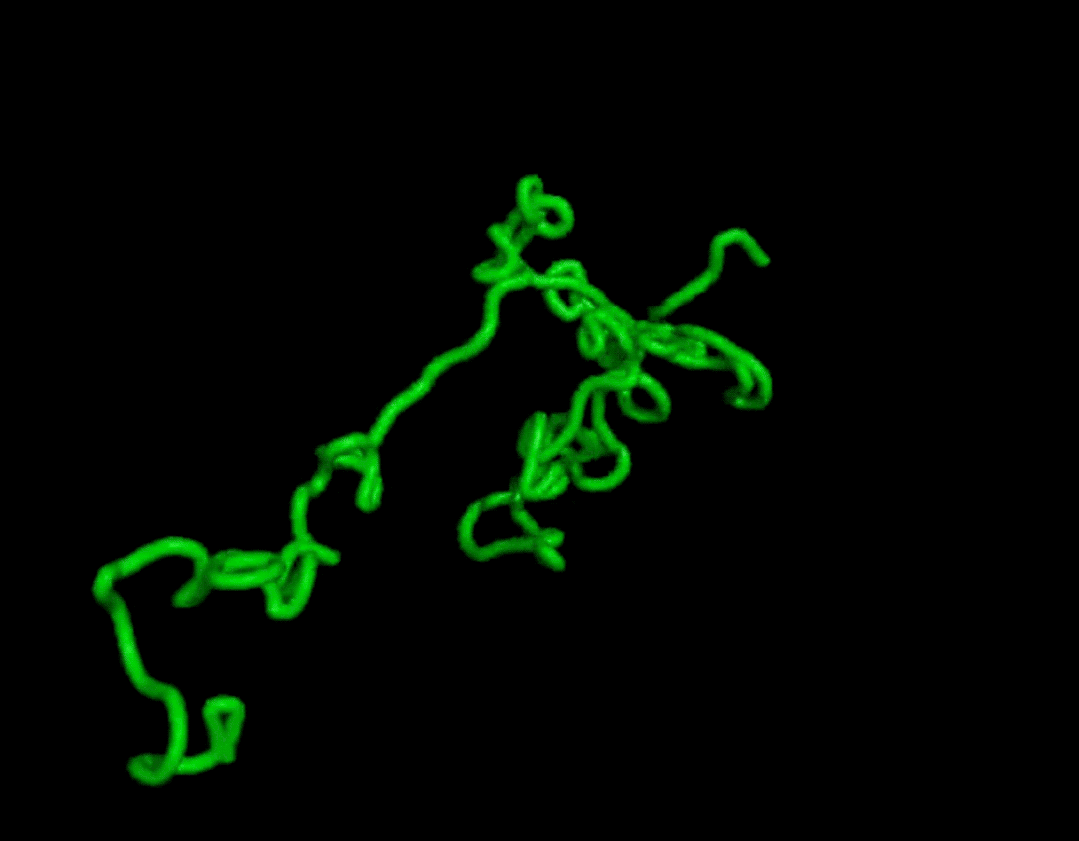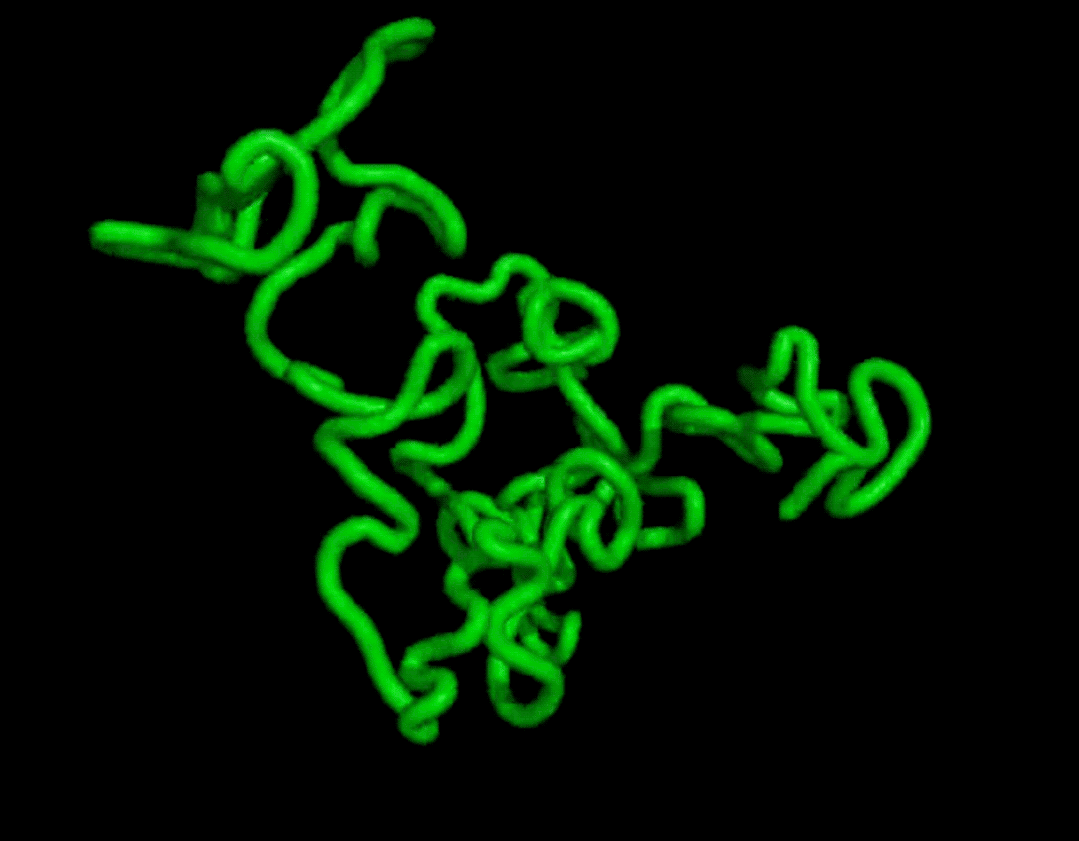
而且結(jié)果在復(fù)雜度和結(jié)構(gòu)上都和天然蛋白質(zhì)有的一拼。

消息一出,不少人都稱贊這個(gè)組合簡直非常妙。

還有人表示:我早就猜到了,生成模型能做的真的不僅是圖像和視頻。
所以,AlphaFold這是可能有新的挑戰(zhàn)者了?

具體是怎么回事?
來看看斯坦福大學(xué)和微軟的這項(xiàng)最新研究成果到底怎么說。

擴(kuò)散模型vs蛋白質(zhì)結(jié)構(gòu)生成
說起研究的初衷,作者表示:
盡管蛋白質(zhì)結(jié)構(gòu)預(yù)測已經(jīng)取得了非常好的成績,但要從神經(jīng)網(wǎng)絡(luò)中直接生成多結(jié)構(gòu)多樣又新穎的蛋白質(zhì)結(jié)構(gòu)仍然很困難。
他們想到用基于擴(kuò)散的生成模型來挑戰(zhàn)這一任務(wù),并通過鏡像蛋白質(zhì)自然折疊過程來設(shè)計(jì)蛋白質(zhì)主鏈結(jié)構(gòu)。
具體來說,就是將蛋白質(zhì)主鏈結(jié)構(gòu)看成一系列連續(xù)的角度,這些角度會(huì)捕捉組成氨基酸殘基的相對方向。
進(jìn)而通過從隨機(jī)、未折疊狀態(tài)到穩(wěn)定折疊結(jié)構(gòu)的去噪就可以生成新結(jié)構(gòu)。
作者表示,這一設(shè)計(jì)不僅可以反映蛋白質(zhì)如何在生物學(xué)上扭曲成能量上有利的結(jié)構(gòu)(how proteins biologically twist into energetically favorable conformations),這種表示的固有位移和旋轉(zhuǎn)不變性也可以極大地減輕模型對復(fù)雜等變網(wǎng)絡(luò)的需要。
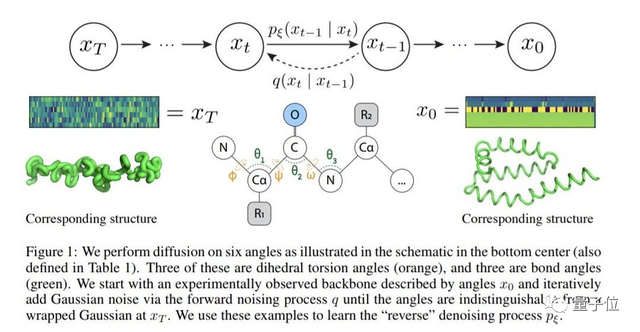
在實(shí)現(xiàn)上,作者僅用一個(gè)簡單的transformer作為backbone就訓(xùn)練出了一個(gè)去噪擴(kuò)散概率模型。
最終證明它可以無條件地生成高度真實(shí)的蛋白質(zhì)結(jié)構(gòu),其復(fù)雜性和結(jié)構(gòu)模式類似于天然蛋白質(zhì)的結(jié)構(gòu)模式。
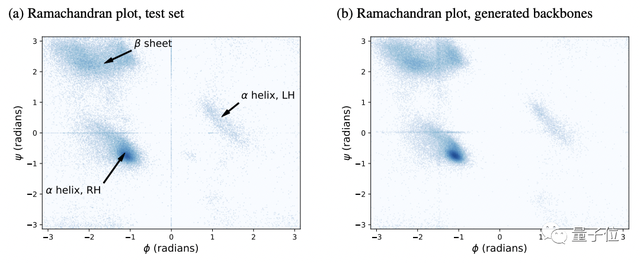
如下圖Ramachandran plot(拉氏圖,專門用于檢測蛋白質(zhì)構(gòu)象是否合理)所示,左右分別為測試集和生成的蛋白質(zhì)主干的(φ,ψ)二面角。
可以看到,三個(gè)主要結(jié)構(gòu)元素、以及一些不太常見的角度組合都在他們用擴(kuò)散模型生成的主干中得到了呈現(xiàn)。
目前,基于以上成果,作者已公開發(fā)布了首個(gè)用于蛋白質(zhì)結(jié)構(gòu)擴(kuò)散的開源代碼庫和訓(xùn)練模型,詳情可戳文末鏈接。
不過,作為一個(gè)初步探索,他們也指明這項(xiàng)成果還存在幾個(gè)局限性,比如:
1、與通常有幾百個(gè)殘基的天然蛋白質(zhì)相比,模型生成的結(jié)構(gòu)仍然相對較短(最多128個(gè)殘基);
2、由于沒有處理多鏈復(fù)合物或配體相互作用,模型無法捕獲蛋白質(zhì)的動(dòng)態(tài)性質(zhì),只能生成靜態(tài)結(jié)構(gòu);
3、將蛋白質(zhì)表述為一系列角度的框架設(shè)計(jì)會(huì)造成一些累積誤差,最終顯著改變生成的整體結(jié)構(gòu)。
最后一個(gè)問題則可以對未來工作提供思路,比如試著用幾何信息架構(gòu)中使用的方法來解決相關(guān)問題。
最后,除了再次刷新我們對擴(kuò)散模型的認(rèn)知,一位畢業(yè)于ETH的研究人員看完這項(xiàng)成果后還大膽預(yù)測道:
未來5年內(nèi),可能漸漸沒有人會(huì)對全新的蛋白質(zhì)序列或折疊感到興奮了。
因?yàn)闈撛诘男旅负椭委熜钥贵w將通過文本提示生成。

對于這項(xiàng)成果,你有什么想說的?
論文地址:
https://arxiv.org/abs/2209.15611
代碼:
https://github.com/microsoft/foldingdiff
相關(guān)閱讀



AI視覺字謎爆火!夢露轉(zhuǎn)180°秒變愛因斯坦,英偉達(dá)高級(jí)AI科學(xué)家:近期最酷的擴(kuò)散模型
AI也會(huì)玩“錯(cuò)覺圖”了



湯曉鷗弟子帶隊(duì):免調(diào)優(yōu)長視頻生成,可支持512幀!任何擴(kuò)散模型都能用|ICLR'24
時(shí)間成本僅產(chǎn)生約17%,可忽略不計(jì)